mgperson2002
Members
-
Joined
-
Last visited
Everything posted by mgperson2002
-
 For anyone interested: There's now an OChemdle game up on the site, with a tournament starting Oct 1. https://organicchemmaster.com/chemdle/signup
For anyone interested: There's now an OChemdle game up on the site, with a tournament starting Oct 1. https://organicchemmaster.com/chemdle/signup -
For anyone looking for an organic chemistry game of the wordle family style, I'm currently working on OChemdle: https://organicchemmaster.com/chemdle


.thumb.jpeg.f7ff55ce93d2f65136c38bd454243511.jpeg)
-
Pinch in and pinch out gestures (using two fingers) are now supported for zoom in and zoom out effects of the molecule. A good example on which to test the new support is https://organicchemmaster.com/MolGen/Molecules/remdesivir?Options=AtomAbbrev Write up of the update is at: http://molecularpathwaygenerator.blogspot.com/2021/09/introduction-of-pinch-zoom-controls-for.html
-
Hi again all. Another quick update. Two new reactions are now supported by the framework of the application: The Benzoin condensation reaction which can be viewed here: https://www.organicchemmaster.com/Molgen/Reaction/benzaldehyde/benzoin?options=Calc,Reac and here: https://www.organicchemmaster.com/Reaction/Rule/031 As well as the Dieckmann condensation reaction which can be viewed here: https://tinyurl.com/5cet2mpv and here: https://tinyurl.com/4tam8kwa In each of the two reactions the first link provides the parameters for a synthesis pathway search (going from molecule A to molecule B) which utilizes that reaction and the second link provides a description of the reaction and a sandbox tool to predict the product of the reaction with a given reactant molecule. I am still looking to create a large team to build this app out to be as useful as possible to both students and chemists. Thanks.
-
A quick update. Hopefully some of the panning issues will be resolved by a new "auto zoom out" feature that will detect if the molecule exceeds the boundaries of the workspace upon its loading. https://www.organicchemmaster.com/MolGen/Molecules/remdesivir?options=AtomAbbrev Remdesivir, among other more complex molecules, will be more easily seen. Thanks again for the feedback. My goal is to one day work with a large team to build this app out to be as useful as it can be to chemistry students and chemists.
-
Right, so definitely not all functionality is available yet on smartphone, but the subset of the functionality necessary for designing and editting a molecule IS there on the smartphone. If there is any confusion about HOW to specifically edit the molecule, there ARE tutorials available (the question icon in the upper right of the workspace). If you are able to, would appreciate any feedback on the tutorial itself. The approach of also adding the pop-up window on the smart phone CERTAINLY is something to be considered. It would definitely introduce all the atom details and click and change to edit modality to the smaller screens. One pretty import consideration, though, is that a small screen often does not have the pixel real estate to both include a reasonably sized workpsace AND the aforementioned pop-up window. My goal for small screen design was to prioritize plenty of screen real estate for the workspace view and editting of the molecule while still offering full editting functionality. Adding the pop-up window was a secondary consideration. That said, I am certainly amenable to suggestions for how that pop up window might work. I'm hesistant to put that pop up window in a place where the user would have to scroll to find it, because I'd like to keep the entire experience of molecule editting contained within the screen size. I'm also a bit hesitant to put in the same pop up style of view/editting, because that would require the user to switch back and forth between viewing the entire molecule AND viewing specific info on one atom. If you have a suggestion for how to maintain the simplicity and practicality of viewing the entire molecule at once while still viewing the detail of an individual atom and being able to edit that atom on a small screen, I am all ears! As for your interface suggestions, LMB instead of hover to select atom: This is interesting. I am open to more feedback of if the volume of users prefer this method. LMB to change bond: Also open to volume of feedback on this. In my mind I wanted to make the editting of the bond type (single, double, triple) or the orientation of the bond (dashed/wedge) to be simple enough to be done WITHOUT the popup window. LMB to pan: This is actually what currently happens. Using the two finger zoom in zoom out as opposed to using the + and - buttons is certainly possible. I am interested in your suggested of select all/select none as well. This could certainly be a future feature to implement. Finally, re: panning molecule view and the scroll top/bottom, do you mean how the molecule workspace is layed out on a small screen? Do you have any other suggestions or preferences for accomplishing this? Keep the feedback coming. Goal is to make this eventually the ultimate o-chem tool, lofty a goal as that is.
-
Thanks again. For now, zoom out has been powered enough to fit the entire Remdesivir molecule in the workspace: https://www.organicchemmaster.com/MolGen/Molecules/remdesivir?options=AtomAbbrev. To further work on is the auto zoom feature which will zoom out to an appropriate level once the molecule is first drawn and the rotate feature. Yes I have a smartphone. Yes I use the website from the smartphone. Yes I am aware there is no hover event exactly as is found in desktop web browsers. The functionality of designing and editting a molecule can actually all be done without the hover event other than creating ions. Though should there be enough interest in implementing the hover features on small screens that can be developed as well. The left click to change wedge-dash notation SHOULD be working correctly on smartphone; what do you mean exactly by "touch display"? There is a chance that it might pick up your finger at a wider sensitivity level than you are expecting. For that I'd suggest zooming in more. If you'd like, you could post a screenshot here and indicate where on the screen you are touching and I can attempt to recreate the issue. Of note, clicking on an atom to change the wedge/dash is actually best thought of as changing the chirality of that atom (of course only relevant in stereocenters) and THEN seeing the change in the visualization of that molecule. In programming terms this means changing the model and having that trigger a change in the view.
-
Thanks @Sensei. Yup do need to work on updating zoom out as Remdesivir is a larger molecule than previously supported. Write up for process of modeling and next steps can be seen here: http://molecularpathwaygenerator.blogspot.com/2020/11/covid-19-ii-remdesivir.html
-
One more update: After a few months the site is now capable of modeling Remdesivir. https://www.organicchemmaster.com/MolGen/Molecules/remdesivir?options=AtomAbbrev. Also working on modeling some relevant syntehsis pathways. Very fortunate the vaccines are now being distributed.
-
A quick update that the site now supports the modeling for the (somewhat controversial) treatment drug of chloroquine https://www.organicchemmaster.com/MolGen/Molecules/chloroquine?options=AtomAbbrev for the novel corona virus. Hopefully planning to model the synthesis pathway soon as well. Of course, if we learn more in the near future maybe that will have to switch over to Remdesivir...
-
Hmm, it seems like williamhakespeare's comment was a copy from an earlier reply hypervalent_iodine. But I appreciate whatever feedback there is! I did finish adding support for the intermediate molecules and reactions used in the Daraprim (pyrimethamine) pathway that was discovered by Australian high school students in 2016: https://www.abc.net.au/news/2016-11-30/daraprim-nsw-students-create-drug-martin-shkreli-sold/8078892 . The pathway synthesis search engine is now capable of "rediscovering" the pathway: https://www.organicchemmaster.com/Molgen/Reaction/1-chloro-4-(2-cyanoethyl)benzene/5-(4-chlorophenyl)-6-ethylpyrimidine-2,4-diamine?options=Calc%2CReac&fbclid=IwAR0qwO493DIEOi-mOy5G7BDn3aZxvSUtMESRlh9bnB602zI_E-llRgAfERE The blog for this update is again at: http://molecularpathwaygenerator.blogspot.com/ Still absolutely a lot of work to be done, and still absolutely welcoming all feedback. Thanks again, Matt Person
-
Thanks for the reply @Avidmind. Again, I am DEFINITELY looking to work very closely with an organic chemist (team of organic chemists) for examples like this to actually draw on their knowledge/experience and incorporate that into the reaction modeling. An update of the progress of the project. I have gone ahead and added support for the user to use the click and select approach for designing a molecule as opposed to the drag and drop approach while in large screen (desktop/non-mobile) mode: http://molecularpathwaygenerator.blogspot.com/2019/10/introduction-of-click-to-select-and.html. The hope is to allow the user an interface more similar to a familar ChemDoodle/JSME style. One further update is that I am currently in the process of modeling the molecule pyrimethamine (Daraprim) and the reactions/intermediate steps involved in one of its traditional syntheses. If enough work could be done on the project that one day power could be taken away from people like Martin Shkreli capitalizing on lack of competition for a certain drug, the project would be highly worthwhile. Finally, I actually am finding myself currently in Australia for the next few days. I will be in Canberra tomorrow (dec 12) and travelling from Canberra to Sydney Friday (dec 13) where I will remain for the weekend. I would absolutely be interested in discussing the project in person with anyone who is available and interested at that time. Thanks again. Matt Person
-
Hi again all. A further update after the addition of some more features. I have added full support for esters in the interface. As such, I was able to go ahead and expand the LiAlH4 reduction rules to INCLUDE reduction of an ester as was previously mentioned missing by @hypervalent_iodine. An example of such a reduction reaction can be viewed here: https://www.organicchemmaster.com/Molgen/Reaction/methyl ethanoate/ethan-1-ol?options=Calc,Reac. Also mentioned was the lack of support for heteroatoms. I have gone ahead and introduced support for nitrogen heteroatoms in particular for this update, with the motivating goal being to support all 5 DNA/RNA bases as well as the medicine allopurinol. A tutorial for how heteroatoms can be added to a molecule can be found here: https://www.organicchemmaster.com/MolGen/Tutorial/HeteroatomMolecule. I am particularly interested in feedback on this approach for adding heteroatoms. Future updates will include other heteroatoms as well. Also of note, the support of allopurinol required, in turn, the support of heterocyclic molecules. Once again, there is still absolutely MUCH more work to be done, and I do sincerely appreciate all feedback. The project blog again can be viewed at : http://molecularpathwaygenerator.blogspot.com/ Thanks all
-
Hi all. An update for those who are following. I have gone ahead and done a re-haul of the interface and specialized/optimized many parts of it for a mobile or small-screen device. Eventually the goal will also be to create an app for ios and android, but for now the site www.organicchemmaster.com will render as it has before on a desktop/large screen device (specifically with screen width of greater than 992px) and will now render in a special mobile optimized mode for a device with a smaller screen. @hypervalent_iodine I did take to mind your suggestion that the draw tool be more intuitive/similar to JSME or ChemDoodle for the mobile version. As such I switched away from a drag and drop approach for adding carbon chains/atoms and towards a click to select and click to add approach. It is my hope to add the bond-line/ zig zag style as an option as well in the near future. @Sensei I did add support for free-radical chlorination. Your search for a pathway from ethane to chloroethane will now be successful. The reaction specifics can be viewed at https://www.organicchemmaster.com/Reaction/Rule/027. There is of course still much more work to be done. The latest updates can be viewed on the project blog at : http://molecularpathwaygenerator.blogspot.com/ Thanks to all who have responded.
-
Appreciated re: your comment of a different design tool for the student than the main one. Will fully take that into consideration. You actually can currently do this. If you click on the molecule name under molecule properties, then enter a different IUPAC name, the molecule structure will change to that name. The basic molecule tutorial describes this: https://www.organicchemmaster.com/MolGen/Tutorial/BasicMolecule . Apologies if I wasn't clear in this case! As long as the IUPAC name is obtainable the structure can be drawn, so this feature can eventually be used for SMILES and CAS numbers. Full enantioselectivity of reactions is indeed something that is planned to be modeled. Currently full stereochemistry support is implemented in the molecule modeling/naming. Some of the named reactions are stereospecific in modeling already, such as this glycolysis step. The thoughts are to include stereospecificity in the rules of a rule based reaction. I had not yet considered convergent syntheses, but I see how those would be valuable. I have some rough ideas for how this might be accomplished using the search engine. That is indeed fair. The process I most reasonably see this project/application taking is FIRST being useful as an educational tool. Then utilizing the knowledge gained and the improvement of the reaction modeling framework (the code backend) to translate that framework to be useful for researcher. You are absolutely right that the second goal will require a much larger team and many more resources and it is indeed more than I can chew. This is the main reason I am actually hoping to build a much larger team, eventually. I KNOW how hard this will be. But I want to see it happen. The first step is indeed to pitch primarily to students. My current thoughts to aid in discovery of a new drug are currently both using the reaction predictor (clicking on the reaction edit pencil will allow you to predict applying that reaction to a given molecule, might change this to a double click anywhere in the row) and including a feature to specially mark a molecule that is either previously unknown, or has other interesting values that is included in a synthesis pathway result. I am absolutely open to more thoughts. I'll take the "I'm a small team" part as a compliment Now it's a free to use web-based service as again, I am looking to gather interest and find others interested in working on the project. Absolutely a tool that is useful for researchers specifically would not follow this model.
-
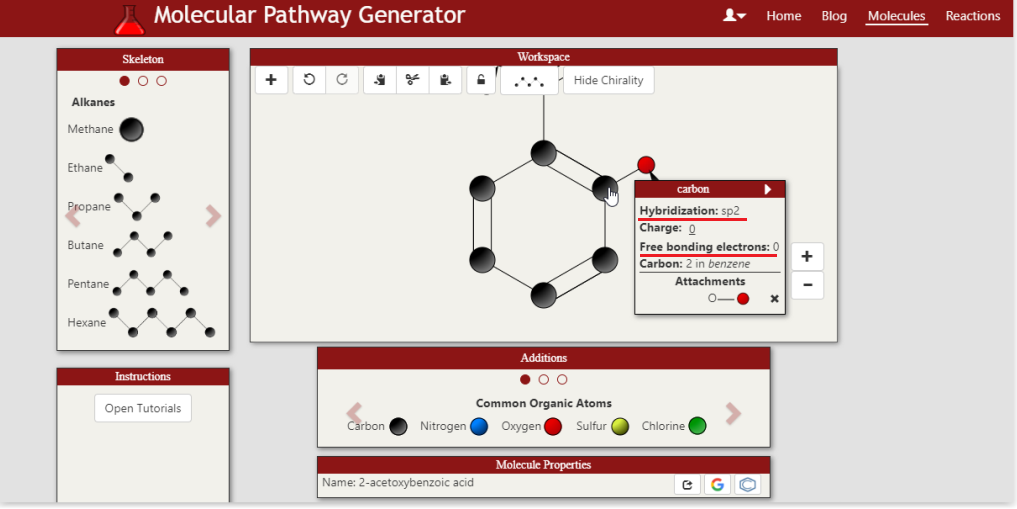
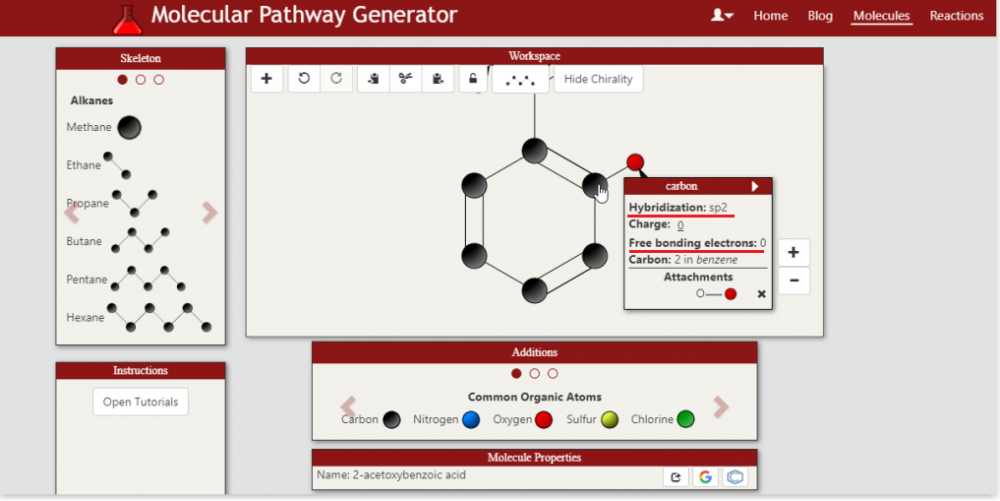
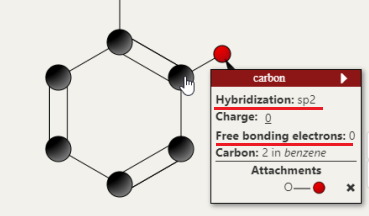
Right, so I will say, the price points that are CURRENTLY listed were actually just sourced as a reasonable starting point to get the lowest cost pathway search function working. Of course the goal again is to make this a dynamically updated, robust system of determining costs of each step in a synthesis. I do appreciate the delivery and tax costs, which I think can actually be incorporated into the search itself with a location parameter. In that particular case you screenshot-ed, the price is actually for the acetic anhydride used to acetylate the salicylic acid rather than the salicylic acid itself (you can see the source by clicking on the reagent name). That said, the price point listed per gram for acetic anhydride is definitely a lot lower, so I have updated the current price point. This price was sourced a while ago, it very well indeed might have been an analytical standard. The pricing and cost generating feature is DEFINITELY one area I want to work closely with an organic chemist or a team of chemists. hybridization and free bonding electrons of atoms immediate updates This refers to the feature of being able to inspect an individual atom in a molecule and view both the hybridization of that atom and its electrons that are free for bonding (currently bonded to a Hydrogen). If you hover over the Carbon in 2-acetoxybenzoic acid that the acetoxy group is attached to, you will see the hybridization as sp2 and free bonding electrons as 0. Thoughts are appreciated about the other areas of the interface. Sounds good! So IUPAC names ARE currently supported and generated on the fly. SMILES can definitely be generated on the fly as a feature to add. As CAS numbers are assigned as opposed to systematically generated from the molecule structure alone (correct me if I'm wrong) this feature will be slightly different as it will require a lookup (either at a cache level or a server pull). In the meantime, the user can click on either the google search or pubchem search icon to pull data about the molecule including the CAS number. Currently Boron chemistry is NOT supported, but absolutely that is another area to add. My M.O. for adding new types of chemistry (heteroatoms, Boron, ester linkages) is I want the IUPAC name generator tool to support ALL reasonable chemicals the user might design first, then introduce that chemistry as a design option. The reasoning is to make the app as robust as possible at each level before adding more. Phosphorous as an atom is not supported, but the Phosphate polyatomic ion is, for such applications as biochemical pathways e.g. the Calvin cycle. Absolutely, agreed still a very interesting project they are doing! The goal of this app is actually to provide a more general approach than a retrosynthetic analysis. The app searches more for a forward synthesis pathway from a user defined starting molecule to a user defined goal molecule. So the starting material is pre-defined, but it's actually not necessarily taken from a set of known starting materials. Nor are any specifically looked for given the goal molecule. The analogy I have come up for is a Google Maps search to find a path from location A to location B, as opposed to finding a lot of known routes to location B from common starting locations. One particular application, and please bear with me as this is purely hypothetical, is a group producing acetaminophen/paracetamol that is used to beginning production with a stock of phenol. For some reason that stock might have suddenly become more expensive, or they might have run out, but they find themselves readily with a large stock of toluene. As they're curious if instead they can produce the acetaminophen from the toluene stock, they enter toluene as the starting molecule and acetaminophen as the goal molecule. This is beneficial if the retrosynthesis might not have identified toluene otherwise as a starting molecule. Again, as this is hypothetical I am indeed making assumptions on availability of materials among other things, but it's meant to illustrate the approach. A further benefit of looking both forwards AND backwards is a different way to discover a new pathway or even a related molecule. So I think the best answer is it's more general in approach, more of a bidirectional search, yet of course can incorporate strategies and algorithms from a retrosynthetic analysis approach. The steps it can theoretically handle in a synthesis are not bounded, nor is the complexity of either of the molecules, but of course the challenge of resources and time becomes higher as these two climb. The search engine of the app is definitely another area that is built to evolve. Heuristics have been implemented to guide the search, as well. To comment on your point of fewer steps preferred, the default synthesis search IS to optimize for the fewest number of steps. Very useful to know. The absolute home run goal of this entire project is ultimately to help discover a new potentially useful drug or even a more efficient synthesis pathway of an already existing useful drug. I absolutely am looking to work with a group of organic chemists. One final note, I am not aware of the etiquette of this forum, but I am certainly interested in communicating over private messaging as well as this thread.
-
Thanks again for the feedback, I appreciate thinking it's a good idea. I absolutely agree there is MUCH more work to be done to make it as useful as it can be. In fact I am looking to get as many resources/people working on the project as possible. In general, a few of the items you mentioned are not supported yet, though of course the goal is to implement support for ALL types of organic molecules. Specifically, esters are not yet supported other than the ester linkage found in an acetoxy group (for example the aspirin (2-acetoxybenzoic acid) molecule ). As they are not, there is no defined reaction rule yet for the LiAlH4 reduction of esters. But yes, that will of course be eventually modeled. As will heteroatoms, which are not yet supported, either. I keep a blog to update when new molecules are supported. These typos are actually VERY easy to fix, fortunately. I went ahead and formatted subscripts where appropriate. The search engine/solver feature DOES allow for protecting group chemistry, eventually. No protecting group reactions (such as Tosylate reactions) are currently modeled, but the search engine WILL allow for those type of reactions. Without getting too much into the specific search algorithms, there is a rule in place such that the search will never consider applying the same reaction to the exact same molecule (under the same conditions) twice, as obviously this could lead to infinite loops (applying and unapplying Tosylation to the same molcule without otherwise altering it). The goal is also to allow the user to control which reactions are considered. There will be logical groupings for reactions such as: toxins, carcinogens, etc, that the user can choose to exclude from consideration. The user will also eventually be able to create custom lists. This functionality can also be used in an educational context, say if an instructor only wants a student to work with a limited set of reactions. Incompatible functional groups will be included in the rule that determines if a reaction can be applied to a molecule. I haven't yet spec'ed out by-products, but I think those can be tracked in the search. Also, of note, the user will eventually be able to define their OWN rule reactions from scratch. I absolutely appreciate the feedback. Yes, per the interface I have indeed considered going with a drawing style more similar to those tools. The drag and drop approach is something I envisioned from the start as a unique approach that also lends well to the immediate, on the fly IUPAC molecule name generation and the hybridization and free bonding electrons of atoms immediate updates. That said, so far feedback has been preferring the JSME style of selecting an atom/bond-line/etc and then clicking the part of the molecule to add to. Moving to this approach will definitely be highly considered. I do think as an educational tool it is important to maintain the inspector feature of the interface so a user can view the properties of any atom in the molecule. This tool can be hidden as well. I did look into the IBM project. From my understanding it is more of a predictive reaction tool. The question it answers as far as I can tell (please let me know if I'm mistaken, I haven't used it much) is "If I combine these chemicals or molecules, what will the reaction and the products be". The question that is asked by the pathway synthesis generator with this project is "What is the most optimal way (cost, number of steps, or total reaction time) to produce this molecule of interest given that I have this (or these) materials available." So it's more of a molecule A to molecule B search than a what happens if we combine molcules A and B. I appreciate your thoughts on number of steps being of most interest. Currently, cost IS calculated from reagent cost. The reagant costs are sourced generally from MolPort, Sigma Aldrich, ABBLIS and TCI chemicals in US dollars. If you click on the reagants in a pathway you can get a link to the source. Right now, these values are static. The goal is to make these sources updated dynamically at regular intervals. Also the goal is to of course take other steps of the reaction into consideration. There will absolutely be an encyclopedia of name reactions. In fact, all reactions in consideration are currently available to view here: http://organicchemmaster.com/Reaction/. What that rule is saying (in plain English) is: 1) Locate a molecule with a Carbon-Carbon double bond. 2) at the location of the Carbon-Carbon double bond, remove the double bond from the molecule and add two Chloride functional groups. One at the first carbon in the double bond and one at the second. The text you see is actually a computer generated translation of what is happening in the coded model of the reaction. So getting that to be readable AND chemically appropriate, as you described, is a work in progress To clarify though, as the reaction is modeled, TWO chlorides are added. One from the "Add" instruction and one from the "Replace". So but-2-ene will be chlorinated to form 2,3-dichlorobutane as predicted/solved for. Thank you again for the feedback, I am absolutely happy to continue discussion. @Sensei Support for Iodination and Fluorination reactions has been added.
-
The main features/goals of the site: A full fledged drawing/design tool to allow the user to build organic molecules following all proper bonding, hybridization, and steric spacing rules. The tool also allows the user to zoom in and zoom out to examine specific areas of more complex molecules by viewing the hybridization, formal charge, electrons free to bond and parent skeleton of each atom of the molecule. Furthermore, the tool provides an instantaneously generated (on the fly) IUPAC name of the molecule the user is creating and convenient links to Google and PubChem searches for that molecule. The user can also type in the IUPAC name to view the structure of the molecule. A future goals is to allow the user to attach custom designed/saved side chains (such as an acetoxy radical) for ease of design. A pathways page to allow the user to explore common and custom organic pathways including common metabolism pathways such as the Calvin Cycle. Specific pathways the user created /discovered will also be stored here under the "My Pathways" sub navigation. Ultimately there is also a tool for a more "admin" level user to approve or reject proposed pathways. A reactions page to allow the user to better understand specific organic reactions (e.g. Friedel-Crafts Acylation and Catalyic Reduction with Hydrogen and Palladium). The user is able to view all associated rules for a given reaction as well as predict what applying a reaction to a particular molecule will yield. The two categories of reactions are 1) reactions by IUPAC name, which involves a reaction specific to a named reactant and product (e.g. Isocitrate dyhydrogenase in the Calvin cycle) and 2) reactions by rule, which involves a rule as described above (convert a carbon carbon double bond to a carbon carbon single bond with a chlorine attached.) A reaction solver feature to allow the user to find a synthesis pathway between both a beginning and target organic molecule(s). This feature can be especially valuable for a student trying to tackle a difficult synthesis problem in which they can't readily find a solution with a web search and ultimately will be useful as a synthesis pathway search engine. An example solution that is calculated is the synthesis pathway from benzene to aspirin (2-acetoxybenzoic acid). This feature allows the user to optimize the synthesis pathway engine search for lowest pathway cost, fewest number of synthesis steps, or shortest reaction time. It also allows the user to specific which reaction sources to search through: the reaction rules, the reactions by IUPAC name, and external sources. Finally, it has an optional "smart search" feature designed to take advantage of a heuristic to predict the most optimal pathway. A home page allowing the user to explore a simplified set of common organic molecules and reaction pathways. This serves as a "lite" version of the site with an introduction to its features. I believe this differentiates from the organic portal OSIRIS property explorer in that it : 1) provides an on the fly IUPAC name generation for the current molecule the user has drawn/designed. This is done locally on the client so does not require a server interaction. 2) Provides a web based application as opposed to a Java app to allow benefits of cloud storage and easier accessibility (eventually when optimized for mobile) 3) only allows user to design molecules that obey proper bonding, hybridization, and steric spacing rules. 4) Allows more detailed inspection of each atom in the molecule for use as a learning tool (including hybridization, free bonding electrons, formal charge) 5) (hopefully) provides a cleaner, easier to use tool including custom attachments the user can define (such as a carboxylic acid or acetoxy radical). From my understanding of both the other features of organic portal and SciFinder (please correct me if I'm wrong) they serve as compendiums of reaction knowledge, research, information, and modeling. The goal of organicchemmaster is to utilize this information to create models of reactions that can be applied to an on the fly, user defined, controlled and initiated synthesis pathway search. Per Reaxys, the differentiation of the search engine is that the user can define a starting molecule and a goal molecule for the search, as opposed to requesting known synthesis pathways for a specific goal molecule. The hope is that this approach will ultimately allow for better discovery of synthesis pathways as opposed to viewing existing ones, but of course that is a very tall task and requires much further work and testing. The question goes from "What are some promising synthesis pathways of this molecule of interest" to "What is the most optimal way (cost, number of steps, or total reaction time) to produce this molecule of interest given that I have this (or these) materials available." In the meantime, the goal of the synthesis pathway search engine is also to provide the chemistry student with a tool to solve problems and learn about reactions (without needing to spend as much money as a tool like Reaxys.) Again, this is a work in progress, so I greatly appreciate all feedback and clarification on any areas I may be mistaken about the other tools/sites you mentioned. I am also interested in potentially collaborating. Finally, I am maintaining a blog of the project at molecularpathwaygenerator.blogspot.com
-
Agreed. This is a work in progress. Bromination actually is currently supported. https://www.organicchemmaster.com/Reaction/Rule/005 You can view a comprehensive list of the rules currently supported here: https://www.organicchemmaster.com/Reaction/# . Fluorination and Iodination are on the future list as well.
-
Thanks @hypervalent_iodine . Yes, it is INDEED not yet optimized for mobile devices, though of course that is a future plan. The discover function there is basically intended as a "teaser" or a very limited functional version of the reaction application feature. The more full fledged version can be found at : https://www.organicchemmaster.com/MolGen/Index . If you're able to describe the typos and other errors I'm more than happy to address them. Thanks @Sensei for testing. I think the answer to your question "So, it's not calculating reaction on-the-fly but searching in database.. ?" is that it actually IS calculating the reaction from ethene to chloroethane, as soon as your click the reaction button, as opposed to doing any sort of database search that would be something along the lines of "Find a reaction that begins with ethene and produces chloroethane". It does not actually run the calculation as soon as you change the first molecule from ethane to ethene though. In the particular case you mentioned of searching for a path from ethene to chloroethane, the reaction it uses can be viewed here: https://www.organicchemmaster.com/Reaction/Rule/019 . This reaction is actually defined as a rule: convert a carbon carbon double bond to a carbon carbon single bond with a chlorine attached. That is, opposed to a more specific reaction like "Ethene becomes chloroethene". As such, this rule can also be applied to convert propene to 2-chloropropane.
-
Hi all, I'm in the process of developing a web based application with the goal of it functioning as a tool for organic chemistry students as well as a synthesis pathway search engine. I would greatly appreciate any feedback. www.organicchemmaster.com Of note it has recently been capable of detecting a pathway from benzene to aspirin (2-acetoxybenzoic acid): https://www.organicchemmaster.com/Molgen/Reaction/benzene/2-acetoxybenzoic%20acid?options=Calc Thanks


.jpeg.6bd9402c226580033ef72af81274ba12.jpeg)