hypervalent_iodine
Administrators
-
Joined
-
Last visited
Everything posted by hypervalent_iodine
-
 thethinkertank has been placed in the queue for spamming the forum with an impressive amount of nonsense.
thethinkertank has been placed in the queue for spamming the forum with an impressive amount of nonsense. -
ginaboyle275 has been banned as a probable spam bot.
-
hipster doofus has been banned following repeated attempts to infiltrate SFN with unsupported nonsense.
-
A controversial opinion, but your logic is sound. I was expecting far more marzipan from you! Disappointing!
-
Who says there isn't?
-
Q-reeuss has been permanently banned for spamming the forum with nonsense.
-
You can definitely hear the difference if you listen to them side by side. Plastic doesn’t give the same smooth, clear tone IMO. It’s not entirely surprising. Some people also say that you can tell a tonal difference between different finishes on trumpets as well, though I don’t know how much of that is actually true.
-
Plastic mouthpieces are very common, but not preferred for normal playing. I have only ever used brass mouthpieces, but my guess is that the plastic would change the tonal qualities of the instrument, which would be why they aren’t preferred. It wouldn’t be to do with stiffness so much as how much the plastic diffuses the sound. You can get metal sleeves to go around the mouthpiece, called brass tone modifiers, and this is meant to improve the sound quality quite a bit, though I have not tried them. Personally, I don’t see that plastic would be too much different to metal once you get going. They warm up fairly quickly.
-
Samatha Priss has been banned as yet another sock puppet of the above. Jojowasaman, Hilary, Intothedarkagain, and Jojo_anderson have all been banned as sock puppets of one another, as well as being spammers.
-
RenaissanceChemist has been banned as a result of being a likely sockpuppet and a definite pest.
-
Right. I think this supports my suggestion of using your draw tool as a separate tool for student learning rather than the main one you use for all of your drawing. I think you might have misunderstood what I meant, or I wasn't very clear. I wasn't talking about generating those parameters on-the-fly, I meant that for your reaction solver or discover function it might be useful to be able to generate a structure from a substance identifier rather than having to draw it in every time. I question how robust this will really be. What you are describing is automated forward reaction planning, which has to rely in some part on automated retrosynthesis and / or the ability for your program deduce a way to build the right complexity to meet a defined end goal. Keep in mind that automated retrosynthesis is something that has only seen real-world success in the last year or two, and relies very much on AI / neural networks / deep learning / other buzzwords. What I am a little confused about is the ultimate goal of this website. The above quote seems to suggest you want it to be used for both researchers as well as students. If that is the case, I think you might be biting off more than you can chew with this particular tool. A research chemist seeking out synthetic protocols would have any number of concerns that I think you might struggle to address. For instance, what if I wanted to make something enantioselectively (this is a very common requirement)? Can it handle only linear synthesis, or could it develop a convergent route (the latter being much more preferable )? I can definitely see how handy it would be to have what you are talking about for a researcher if it were fully fledged and capable of handling a lot of complexity and a lot of reactions, but at the end of the day I would really question whether or not this is a goal you can reasonably achieve within the frame of a free-to-use web-based service with a small team behind it (I assume this is the case, apologies if not). Moreover, I wonder if by pitching this for students and researchers, you are spreading yourself a touch too thin and venturing into the realms of doing too much at once. Everything else in your website to me seems like it is geared largely towards education, so the reaction solver / design function seems a bit...out of place I guess. That's just my 2 cents. I haven't really seen anything in your website that would lend itself to this goal. Could you elaborate?
-
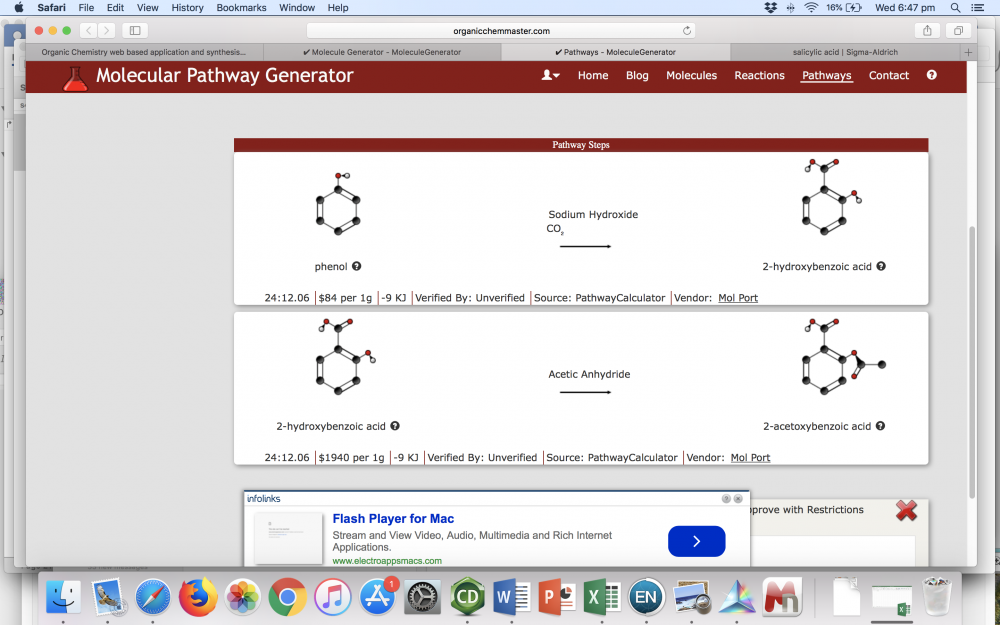
So, with Sigma you can get wildly different prices depending on what quality / grade you are okay with. For synthesis, you generally do not need analytical grade chemicals. I bring this up because when I went back to have a look at your reaction solver, I had a look at the suggested pathways for the example you linked to. This is what I get: The 2-hydroxybenzoic acid in the second example is aka salicylic acid. When I saw the price listed I immediately went and checked Sigma because there was no way something as common as salicylic acid costs $1940/g. When I check Sigma, I can buy 3 kg of the stuff for $US200. The only thing I could find close to the price you list was an analytical reference sample (50 mg for $US123), but no one is using an analytical standard to perform synthesis unless they have an absolutely wild disregard for budgets. One of the reasons I asked about where you were basing costs is because if you live somewhere like I do (Australia), you have to factor in a lot of other costs for delivery and tax. This is not really a problem if the intended users are students and not researchers of course. Firstly, I don't understand the bold statement. Unfortunately, the tool you have now is not very easy or intuitive to use (IMO), which I think should be a key focus over simply making something that is different. I think that the tool that you have with the on the fly IUPAC name generation would be good as a separate tool. As in, don't use it as the primary draw tool for your reaction solver or whatever, just use it as a specific tool for students to learn about IUPAC nomenclature. I think having something that creates a name as you build a molecule would be very good for that, but I can't think of a single time when I have been using SciFinder or similar to look up a reaction and have needed a IUPAC name to be generated as I construct a molecule. Another comment I would make about it is that you should integrate a way for a user to freely move around atoms. I was using the tutorial just before to make aspirin, and when I added an ethane to an oxygen atom, it added it in such a way that it overlapped with the phenyl ring. Upon trying to add the carbonyl oxygen, I instead got a carbon that appeared triple bonded to my original oxygen, and there was no way for me to change it without deleting it. I would also recommend you have a function to convert other identifiers into structures, like CAS numbers, SMILES, or IUPAC names. Finally, more atoms! Boron chemistry is pretty common for example, as are molecules with phosphorus, but you have neither as an option. Yes, you are correct. I half misremembered what it was and didn't look it up when I posted. Still an interesting project! From what I can tell, you are essentially automating a retrosynthetic analysis on a compound back to some pre-defined starting material, and then displaying that as the equivalent forward reaction steps. These forward reactions are shown as A --> B with the main reactants that one might use. Before I comment further, could you tell me if this is this correct? What level of complexity do you anticipate you will be able to handle? As in, how many retrosynthetic steps could it do, and how complex can the molecule be? I happen to work as an organic chemist in a group that does a lot of drug discovery and natural product total synthesis. In total synthesis, the things that we really aim for are (as I mentioned) shorter steps, higher yields, and fewer chromatography steps. The big thing is shorter routes and higher yields. Cost of the reagent is obviously critical also, particularly in industry, but having few steps, high yields, and easy purification are often the things you look at first.
-
Ahmed Torah has been banned permanently for posting a lot of preachy nonsense.
-
Okay, I have looked at it briefly on my lap top. It is a good idea, but I think you need to invest a lot more time into it before it becomes useful. Before I get too much into it, I have to say that I don't like the draw tool you use. It's very limited and not super intuitive. I tried to step through the tutorial to draw ethanol, but could not get it to add any heteroatoms? Could you not integrate something like JSME or Chem Doodle? These are more complete in terms of a draw tool, and they use the more common bond-line / zig zag style of drawing. I think JSME is free to use as well. You may be interested in a recent project launched by IBM. It essentially performs the task of your reaction solver but uses a data driven AI approach, which I think is very elegant. Is this what your tool does as well, or do you use a different method? Out of curiosity, how do you calculate cost and reaction time? Does it take in to account purification and work up steps, or just the reagent cost (and if so, where do you source this and what country are you basing it from?). Reaction time is quite variable doesn't always translate across different molecules very well. I think most chemists would be more interested in limited number of steps, as this invariably leads to a reduction in cost and time. I don't think that trying to sort by some arbitrary cost or 'time' value would be that helpful. Another question I have with the solver feature is how it accounts for possible by-products and incompatible functional groups. Does it incorporate protecting group chemistry? Does it allow for the user to refine the types of reactions used? As for typos. Firstly, I would recommend sub-scripting in your chemical formulae where appropriate. You also use a lower-case k for K2Cr2O7. The wording you use is also very confusing in some places. I don't have time to go through it all, but I recommend hiring someone to proof read it. For example: This is the action for Cl2 addition to a double bond. I honestly do not understand what it is saying. Is it saying you only add one chloride to the double bond? That seems to be the interpretation, but it is incorrect if this is the case. I also don't like the use of the word replace. You use it when describing oxidation of alcohols to aldehydes, but this isn't really a good way of describing it IMO. Another thing I noticed is that your LiAlH4 reduction rules do not seem to account for esters. Will you have an encyclopaedia of name reactions?
-
So what does it offer over other websites like organic portal, SciFinder, or Reaxys?
-
It doesn’t seem to be well optimised for mobile devices would be my first comment. My second one is that I don’t really understand what the website is trying to achieve exactly? As a synthesis pathway engine it is not particularly useful as-is. From what I could see you only seem to be able to optimise from a very limited selection of molecules using biochemical pathways? Perhaps this is just because I am viewing it on my phone. The discover function again is very limited, and contains some typos and other errors.
-
! Moderator Note coffeesippin, Science =/= the opinion of one scientist. What you have presented is not science, nor evidence for anything that you claim. In fact, it is little more than another attempt at preaching. Staff will not continue to warn you about this. We will ban you permanently if it keeps up.
-
Suzie has been banned as a sock puppet of Olin (who, incidentally, was a sock puppet of Menan).
-
coffeesippin has been suspended for one week for abuse behaviour and preaching.
-
FreeTheGenius has been permanently banned after a particularly abusive and unwarranted PM.
-
Endercreeper01 has been permanently banned for persistent violations of soap boxing, and rule 1 c.
-
! Moderator Note I think we’re done here.
-
For the sake of transparency, we have decided to announce who has been added to the groups mentioned in the OP. Currently, they are as follows: 0-point (aka Curmudgeon) xyzt (now banned) et pet TED888 Conjurer (added Feb 8 2020) 2-point (aka Malcontent) Itoero John Harmonic (now banned) I will continue to update this post as necessary.
-
Vvi has been banned for an abusive tirade about numbers. O Gor has been banned as a sock puppet of OlegGorokhov
-
In previous threads, staff have mentioned that we have on occasion curtailed or removed the ability for people to use the reputation system. In previous versions of the forum software we were able to limit positive or negative reputation limits separately. This meant that the impact of people who wished to use the reputation system to target specific members was always low, and staff could easily reverse it. This no longer being the case means that the system is more open to being abused by people wishing to use it as a form of personal attack. Thankfully, we have had very few cases where staff have had to intervene. For those cases where we have had to do something, admin have created two new member groups with reputation point limits set to either 0 or 2, which limits the use of both positive and negative rep points.