Dan_Ny
-
Posts
64 -
Joined
-
Last visited
Content Type
Profiles
Forums
Events
Everything posted by Dan_Ny
-
Is it possible to design a smart-toxin?
Dan_Ny replied to Hypercube's topic in Biochemistry and Molecular Biology
Yeah, another example for a DNA binding (and inhibiting helicase in DNA replication) is the mitomycin family. But this area of DNA bonding is still quite new and a lot of work needs to be done - since the targeting site of these molecules is the minor and major groove of DNA, in principle it should be possible to design a "small molecule" for a specific sequence - but, if that sequence is longer than a few base pairs, the inhibitor of helicase must be damn complex and long - and able to wrap around the DNA a couple of times... If I had 100 years to synthesize some of these molecules, I would do it, but there must be an easier way It would be cool if we could desing a few enzymes which can be somehow produced on demand and which synthesize such an DNA-intercalating toxin in vivo to adress Mr Skeptic's fragility problem.... Could you post some references? That sounds damn interesting. -
Even though water is very often our worst enemy, it can have it's uses sometimes...
-
Hm. Is there no way to elucidate the absolute configuration of a natural product? Once the NOESY gives you the relative configuration of the stereocenters, the assignement of one absolute stereocenter in your molecule should be sufficient to elucidate it's overall stereochemistry (I hope I got that right). Why not simply modify your target with a chiral reagent, take the NMR of that and assign a stereocenter - knowing the enantiomer you want now, you can take stereoselctive reactions to build up your molecule instead of going through the racemic route. Without the trouble of carrying through several diastereomers, you prepare only one... But when it comes to chiral catalysts or auxiliaries, I totally agree that generally you first want to quick (i. e. quick, cheap + racemic) check the feasability of the route and after that go for the enantioselective variant. And what about chiral NMR sovents? Do they exist? Can they be any help in regard to which enantiomer of a nat pdt is at hand?
-
Mixed claisen condensation: diethyl carbonate w/ NaOCH2CH3
Dan_Ny replied to Genecks's topic in Organic Chemistry
Indeed, it is more stable - but it is not consistent with your observation that carbamides are more stable than carbamates, is it? And still, the question remains, if "carbonates" are also more stable towards harder nucleophiles. I will look that up tomorrow, I think. -
Mixed claisen condensation: diethyl carbonate w/ NaOCH2CH3
Dan_Ny replied to Genecks's topic in Organic Chemistry
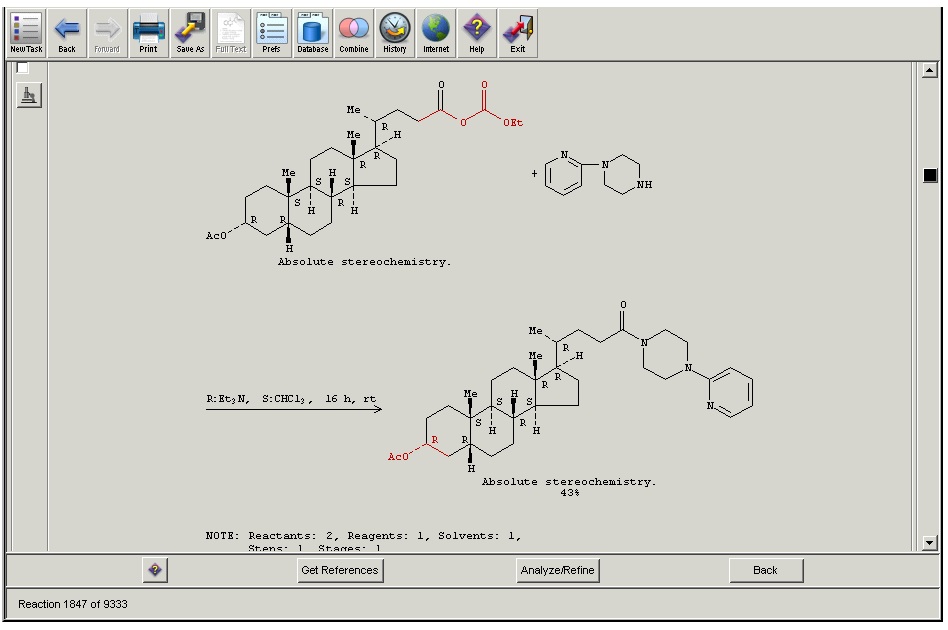
No need, I've already looked up scifinder. Bioorganic & Medicinal Chemistry Volume 16, Issue 18, 15 September 2008, Pages 8737-8744 (See attachment) So we have here a secondary amine as the nucleophlie which has the choice between the carbonate and the ester functionality and it goes for the ester carbon. In this case the reactivity of the ester really is higher. Well it is fun to talk about, though. Because it could to a certain extent actually be the type of nucleophile that chooses between its electrophilc target. The electron-poorer carbonate functionality might be the harder electrophile and therefore more prone to attack by something hard, lets say grignards or organolithiums, while the "electron-richer" ester is softer, making the amine, a soft base attack itself. (((This is a rather heretical remark: It is quite fun that in Chemistry we will always find explanations for everything - might it be the HSAB concept or inductive effects oder hyperconjugation or whatever effect overriding the other... In the end one can "explain" everything and therefore - nothing. Well, of course I'm exaggerating a lot... But sometimes I get that feeling...))) I definitely have to find an example where BuLi has to attack that kind of structure. I would guess, though, that it might attack the carbonate, rather.
-
Mixed claisen condensation: diethyl carbonate w/ NaOCH2CH3
Dan_Ny replied to Genecks's topic in Organic Chemistry
Ah, right, of course. I wouldn't call it shielding though, it should be rather a transfer of electron density (well, you said it - conjugation). Just wondered about the reactivity in the first place because I have never come over a nucleophilic attack to a carbonate like this. There may be something in the literature, though. But if they are less electrophilic than, lets say, amides, it is clear why I've never seen it before. -
Mixed claisen condensation: diethyl carbonate w/ NaOCH2CH3
Dan_Ny replied to Genecks's topic in Organic Chemistry
Yupp, that's what I think, too. -
There has been some tumult about Maoecrystal V (having three vicinal quaternary streocenters). And the diels-alder reaction in the final step really is beautiful... Gonh, J.; Lin, G.; Sun, W.; Li, C.-C.; Yang, Z., J. Am. Chem. Soc., 2010, 132 (47), pp 16745–16746 Actually, there have been other, interesting approaches to this crowded molecule: Baitinger, I. ; Mayer, P.; Trauner, D. Org. Lett. 2010, 24, 5656-5659. (As well as many other papers...) But nothing beats a good Diels-Alder. Up to 4 stereocenters in one step, perfect atom economy etc. pp.. Is there a better reaction?
-
Wow, for the next 1000 extremely difficult NMR questions I know myself in good hands now. And I WILL post them. No, seriously, brave job from Horza2002 and hypervalent_iodine!
-
Yeah, like Horza2002 said, you need to take some stain and the usual silica tlc plates. It is the most simple thing on earth. I don't know the exact properties of sugars on silica, but try mixtures of methanol and DCM or ethyl acetate/DCM or ethyl acetate/hexanes as eluents (mobile phase) or something in that polarity range. A good way to find a good "mobile phase" (with the desired retention factor) for your compound is just to make 5 different solvent mixtures at once, prepare 5 different tlc plates at once, put them in there, run them and stain them (spares time). Take the best solvent mixture or change polarity again after that. The sugars themselves should not be visible in the UV (if you use the nomal tlcs, that is), but KMnO4 should definitely stain your sugars in high enough concentrations. Otherwise, have a look at Horza2002's list of stains...
-
Mixed claisen condensation: diethyl carbonate w/ NaOCH2CH3
Dan_Ny replied to Genecks's topic in Organic Chemistry
Yeah, that makes sense. How electrophilic are these Dialkylcarbonates compared to esters? -
Sounds convincing. Especially that part about my mind. It would be quite unfortunate, then, not to be a good organic chemist, right? Well, only the recognition of the undesired diastereomers in question might become a problem. Imagine them to be unseperable by silica... same mass in the LCMS, of course, and those NMR spectra of complex precursors - I need quite a big amount of endurance for those sometimes... But yeah, I agree, the best way to solve that is to think by yourself about what might happen to your molecule in a specific reaction.
-
I think it depends on the solubility of your aldehyde and its stability towards aqueous conditions (I am not sure about hydrate formation and when that occurs and if it is revesible etc.). In general, to get rid of DMSO is the following (if your product it is considerably more soluble in organic solvents (like ethyl acetate, DCM, diethylether or others)): Take the reaction mixture after completion of the reaction. Add water (or Sodium Phosphate ph7 buffer). Extract a couple of times with a volatile organic solvent (DMC, or one of the above). Wash the organic layer with water two or three times (take 10 times as much water as the amount of DMSO you had in the reaction!!!). DMSO is more soluble in water than in organic solvents. It will be gone. Evaporate organic solvents. Take a crude NMR, you know, column and then stock solution in benze, freezer. The usual procedure. Best, Dan
-
Hi all, since I have a lot of questions about what is commonly referred to as "Total Synthesis of Natural Products", this Topic is for questions about it that might occur to anybody at any stage of their organic synthesis efforts. My first question would be this: Why is the first synthetic aproach towards a target molecule always a racemic approach using racemates as starting materials? Is it not possible that the diastereomers, thus formed, will react quite differently giving misleading results and yields? Thanks to everybody in advance for their help, answers and questions. Dan