Suxamethonium
-
Posts
183 -
Joined
-
Last visited
Content Type
Profiles
Forums
Events
Everything posted by Suxamethonium
-

Potassium Hydroxide Separation
Suxamethonium replied to elementcollector1's topic in Inorganic Chemistry
"What will end to happen is that calcium sulphate will ppt out on the surface of the (fairly insoluble) Ca(OH)2 and stop any further reaction." That is why it is being continually stirred and sulfate added slowly- it's less likely to form a suitable enough coating to hinder the calcium hydroxides solvation- at the very least you would get SOME KOH which is much easier to seperate from the Calcium ions. As for the heat released- add it slowly its not exactly rocket science- and there is sufficient water to absorb this heat without boiling so a fire is highly unlikely in even the worst case scenario- NB. have you never made limewater before? Also leaching bon fire ashes with water is still going to contaminate your solution with other cations (including sodium) so I don't see how this is 'easier to separate' than the original mixture, except that the ions envolved aren't quite so interesting. -
Sorry, it sounded like you were going to use it to neutralise the nitrating reagent- You could do just nitric acid, but this requires higher temps or activated reactants (as the electrophilic nitronium ion isn't produced in nitric alone). What you are doing is essentially an esterification just using an inorganic acid- so yes nitric will work, but nitric/sulfuric mixtures are better at it. Alternatively try sulfuric and potassium nitrate mixtures. Also, neutralisation of the gun cotton after the reaction is mostly to make it non-corrosive- It does stop any continuing reaction, but such reactions wouldn't continue long anyway- especially if you washed even just with water. From memory cotton has 3-4 nitratable positions per monomer. I'm assuming you only want mono-nitration. The best way to acheive this is just to keep the reaction mixture cold (on ice for 15mins or so).
-
Potassium Hydroxide Separation
Suxamethonium replied to elementcollector1's topic in Inorganic Chemistry
I would imagine that the solvation gradients are significantly different for KClO and NaClO (considering that other chlorates are similar in such a way)... so you could ppt one or the other and be left with either KOH or a mixture of NaOH and KOH. If KClO ppts you could always convert it to the hydroxide then seperate from the contaminant of KCl. Edit: If you want a displacement reaction: K2SO4 + Ca(OH)2 --> CaSO4 + 2KOH The trick would be to get a large beaker of distilled water and stick it on a stirrer plate. Add sufficient calcium hydrodixe or oxide and let it stir for a few mins. Keep it on stirr and add saturated potassium sulfate slowly until stoichimetric ratio is reached. Calcium sulfate will precipitate out quickly and eventually replace the calcium oxide floating in the beaker. -
What kind of substance should I use..
Suxamethonium replied to NNenov's topic in Inorganic Chemistry
No worries. Um- white spirit here (australia) can also be like a shellac kind of solvent. Still experiment with it, but I don't know if it will give you the same turbulent effect as ethanol/methylated spirits. As for opaqueness.. well, now things ae getting harder. You could probably work wonders with colloids, but its pretty involved chemistry requiring 'just the right amount' lol. nanotechnologist or combination with a specialist physical chemist is probably the best area to find helpful details on such chemistry. You could try just using LOTS of food dye? But eventually it would dissipate. Alternatively mix large amounts of red, green/yellow and blue food dyes. This will absorb all the wavelengths effectively creating a deep black that will persist. -
I feel that last equation is wrong, but the process/products is correct. Atmospheric oxygen is doing the oxidation (I think of Fe(OH)2 to Fe(OH)3 which is even less soluble)- this is why it's 'purifying' the Mn(OH)2- which somehow dissolves and is left in solution with the chloride ions. It looks like it could be a complex combination of reactions, which my best guess is something along the lines of: Fe(OH)2 (s) (miniscule amount)> Fe(OH)2 (aq) Fe(OH)2 (aq)+ 0.5O2 +H2O (or + H+) > Fe(OH)3 + -OH (or + H2O) Fe(OH)3(aq) (large percentage)> Fe(OH)3(s) Now there is 'more room' for more Mn(OH)2 and Fe(OH)2 to dissolve in miniscule amounts once again (even though they are 'insoluble' a miniscule ammount dissolves enough to get a slow reaction- these are both more soluble then the Fe(OH)3 which is why the reaction is driven forward- by Le Chateliers Principle). Overall, all of the Fe2+ becomes Fe3+. And as such all the iron recipitates out as iron (iii) hydroxide in preference to the Mn2+. If the Fe3+ somehow sequested all the HO- present in solution then only Mn2+ and Cl- would remain in the solution. You also have a significant amount of Cl- present and the video isn't exactly clear, but I imagine it is slightly acidic initially- otherwise the alkalinity created by the reaction would cause more precipitation. In such a case maybe the OH ions in the Mn(OH)2 can be displaced by the Cl- ions? Like I said, I'm unsure, but it is definately not a single reaction occurring. By comparison the second video is straight forward. I would use the KHSO5salt if you can get it, if not use a similar salt like NaHSO5 or NH4HSO5 or even peroxysulfuric acid. If still no luck try the dication salt mixed with sulfuric acid or as a last resort try an acidic solution of S2O82- salt (much less likely to work). I still feel hydrogen peroxide will not work effectively- although it seems Mn(iv) wont be reduced by the peroxide like Mn(vii) it still directly catalyses its decomposition. The reason is because even without the loss of Mn(iv) product, as more of it is prodcued the rate of hydrogen peroxide breakdown will increase thus decreasing the rate of the usefull reaction significantly (and increasing the amount of hydrogen peroxide needed, and heat generated).
-
What kind of substance should I use..
Suxamethonium replied to NNenov's topic in Inorganic Chemistry
Yes. You will need to experiment with the idea a bit to get exactly what you want- but it is cheap and easy. You need a shallow pan (whatever colour/finish that works best with your camera is fine). Then pour a little water in the pan. Add methylated spirits. The alcohol in the spirits will produce a significant turbulent effect. You can experiment with food colourings, temperatures (say heating the water or spirits before hand), amounts/depth. You can even use other alcohols like isopropyl alcohol. Beware of the flammability risk, and avoid using food pans- but you could always just wash thoroughly afterwards. The other option is the large scale density differential effect. You'll need a large tank of cold water and a jam jar or similar. Boil some water, colour it and pour it into the jar- put the lid on and submerge the jar in the tank. Slowly remove the lid from the jar and the hot coloured water will rise to the surface in various turbulent patterns (this is pretty much the same as the tea/coffee and milk effect- albeit reversed). If you wanted imissable liquids (like oil and water) you would probably need to mechanically stir or agitate the mixture to get your effect- or rely on significant temperature differencials. Again, you would need significant experimentation to get the look you want. If you want to increase the vicosity of aqueous solutions an easy way is to add sugar or glucose syrup- this also increases the density. Increasing the density of oils/solvents is harder. You might want to try solvation of solid fats (lard or butter)- but I am not sure how great that would work having never done it. Happy experimenting. -
No it can't remember what it was before, but thats not to say something won't crystalise differently at a different pH. This is probably much more prudent in organic structures, but whilst unlikely to change anything you could always try and see if your recrystalisation is any different in acidic conditions- it wont really matter what acid you use (provided it's a simple acid and not something thats going to completely complex with your boric acid) HCl or H2SO4 should be fine- may as well try different concentrations whilst you're at it.
-
So it is proposed that: Mn(OH)2 + 1/2O2 --> MnO2 + H2O This would probably work- but I doubt it would be spontaneous. You would surely need some energy if not just to drive off the water. Manganese oxides structures tend to be fairly variable at the best of times, you're probably more likely to get something like (and I just pulled this formula out of my head to illustrate the point): Mn(OH)2 + O2 --> MnO(OH)2 or even worse a co-complex like MnO.MnO(OH)2 ... Going straight from (ii) to (iv) is also less likely to be spontaneous as say (ii) to (iii) to give the very common brown Mn2O3 (also forms messy complexes like MnO.Mn2O3). You're best bet is probably to find something unusual that Mn forms a soluble salt with (possibly ligands would be a good start), or go the opposite way and see if ligands like EDTA are non-effective for Mn ions (in which case EDTA could stop Fe precipitating with the Mn). Then worry about oxidation states. As for the H2O2 and NaOH. Hydrogen peroxide is not a good idea. Mn salts catalyse its decomposition, and salts (iv) and higher are often reduced to (ii) or (iii) salts. Also, you wouldn't add the NaOH until the end of the reaction or you would precipite everything and get no reaction at all.
-
Um. There is one major issue here which I will get to later- but first lets address the other points. For commercial HCl to be green it is either coloured specifically with some organic dye. Or it has very significant impurities (probably of the transition metals). However Iron in concentrated hydrochloric acid is yellow due to the formation of the FeCl4-complex. It could be another transition metal though- maybe copper or nickel- although these too will be yellow in HCl of sufficient concentration. Iron in the battery depends on the battery- I happened upon some batteries where the case was iron (steel) with a zinc coating on the inside, or some with the electrode cap being iron- these could corrode into the battery over time. Was the ingredients list specific for that battery, or just generalised? Also, it may have only listed hazardous or active components in which case iron might be listed with 'other non-hazardous contents'. Now, from my experience with trying to extract transition metals from batteries. I personally got the green colour myself dissolving cadmium batteries in conc. HCl. I put the green to the presence of nickel (it went yellow when I replenished the acid), and cadmium. I concluded manganese would not impart any colour to the solution and this is the main issue in your method. The HCl is oxidised by the MnO2 into Cl2 H2O and MnCl2. Changing pH would then yeild Mn(OH)2 not Mn(OH)4 the latter of which would dehydrate to MnO(OH)2 and on heating finally to MnO2. Instead you will need to reoxidise Mn(OH)2 or change the original acid so as to maintain the +4 oxidation state (sulfuric comes to mind). Finally- acetates of manganese are soluble (In fact- most simple organic acids will have soluble metal salts- even lead!). Manganese (iii) acetate can spontaneously disproportionate into manganes (ii) and (iv) acetate- this may be an efficient way to reoxidise you manganese by utilising a reagent to oxidise (ii) to (iii) which disproportionates back to (ii) and some (iv), the (ii) can then react with the oxidising reagent again.
-
Sorry. Wouldn't Cu2S be copper (I) Sulfide? CuS would be copper (II) sulfide.
-
So, when you did the recrystalisation before, letting the solution cool- you said you got the right sort of crystals, but they didn't compact to form the flake? What I feel is likely to have happened was that you got a bigger crystal (same shape and texture, but the increased size wouldn't compact as easy). You could try to induce smaller crystals by precipitating them from solution faster (i.e. rapid cooling (on ice) and scratching the bottom with a glass rod to induce precipitation). As spectulation, I would imagine industry would cool and induce precipitation in such a way because it is easy to do, and quickens the overall process- this might be why they achieve the smaller, more compactable crystals.
-
Yeah, sorry. I didn't actually check if you were American or not when I implied that- For that you have my apologies. However, just because a fuse is not American doesn't make it Chinese by default. Also- I was under the opinion (I could be wrong it's not exactly my area of interest) that Japan were the elite in pyrotechnics- I was reading in a newspaper around the time of the 2000 NYE which claimed the highest end pyrotechnics employed originated in Japan. Also, being in Australia, if I was so inclined to make explosives of any degree, specifically looking for American fuses as opposed to any countries high quality fuses seems like a waste of effort and time- so maybe it would be more effective simply to invest in good quality fuses regardless if they are American or not?
-
No offense intended, but I get this feeling that American's seem to feel they do everything the best. So I was just wondering the evidence you are basing that statement on.
-
Im confused? Could you not just make hydrogen and oxygen in different chemical reactions? Then you don't even mix them in the first place. Hydrogen - acid and metal or NaOH and Al. Oxygen - Hydrogen peroxide and catalyst. Mixing them can be dangerous (explosive) though, so be careful if you plan on doing such- or preferably don't do it.
-
My first question is "why is over nitration bad?" After that my second question is "So, if what you propose doesn't work then you will still get excessive nitration. Are you still prepared to carry out said experiment knowing that risk?" My third question is then "Ok, so how are you going to know when it is nitrated sufficiently?". Also, just a tip- adding baking soda to concentrated nitric acid is not a wise idea.... its going to take a lot of baking soda and I would not be surprised if you ended up nitric acid all over your work-space (YAY! Volcano!!). Also unless you neutralise it quickly the added temperature will increase the rate of nitration.
-
Last time a performed a thermite reaction in ceramic it shattered. You could try a heavy walled ceramic- but you'll probs need to go with something like cast iron (and hope the reaction doesn't exceed 1200 degrees C. Mg and Al should be reactive enough, at the very least you will get some cerium (but you may have to use excess Mg/Al).
-
Calcium chunks are usually stored in an air tight container. After long storage you end up with calcium oxide/hydroxide/carbonate instead- but it takes a while- filling the container with argon or nitrogen would prolong the life considerably- but its not necessary. As for the reaction, calium chunks wont work very well (if at all). You would be much better off using powdered metal, noting that Mg or Al are readily available in such form. Alternatively you could try to melt the calcium (probably once mixed with the cerium oxide) to start to reaction- you'll probs need the blow torch (approx 850C) and a container that can withstand to reaction temp.
-
You could try Ca(OH)2 and K2SO4 to yield the KOH in solution... but then you would have to dry to K2O and rehydrate just enough to get KOH again (if you want dry KOH). If you're going to do that you may as well just decompose K2CO3.
-
Start here. http://www.chemistry.ccsu.edu/glagovich/teaching/311/content/alkene/reactions/alkenerxn2.html Then google 'millons test'. Start by reading the (very short) wiki article, and then from what you learn there google some of the key words and see if you can't work out its importance on your own. If you get stuck i'll give you some more help.
-
How can i know what kind of gas i have in my jar?
Suxamethonium replied to mScientist's topic in Applied Chemistry
lol... there are lots of tests- some more affordable than others. Colour, smell, and density are all fairly easily determined. As is pH. You could test for oxidising properties (O2, Cl2, N2O for example) or flammability (H2 or HCs for example- if so, run an ms on it). You could measure its UvVis absorbance to compare curves with known (suspected) compounds. Various gas tests like CO2/limewater tests could be imployed. Depending on what the application is, its only likely to be a select few gases- it shouldn't be too hard to deduce it from logic and trivial data alone. -
There are so many possible reactions to do that eventually end up in alcohols/diols, aldehydes, ketones, alhp-beta unsaturated ketones etc etc. You could even make chlorides or nitrides and hydrolyse to get such products.. Do you want anything specific? Or just general reactions (hydrogenation, epoxidation, etc)?
-
Question About How Catalysts Work
Suxamethonium replied to The Chairman's topic in Applied Chemistry
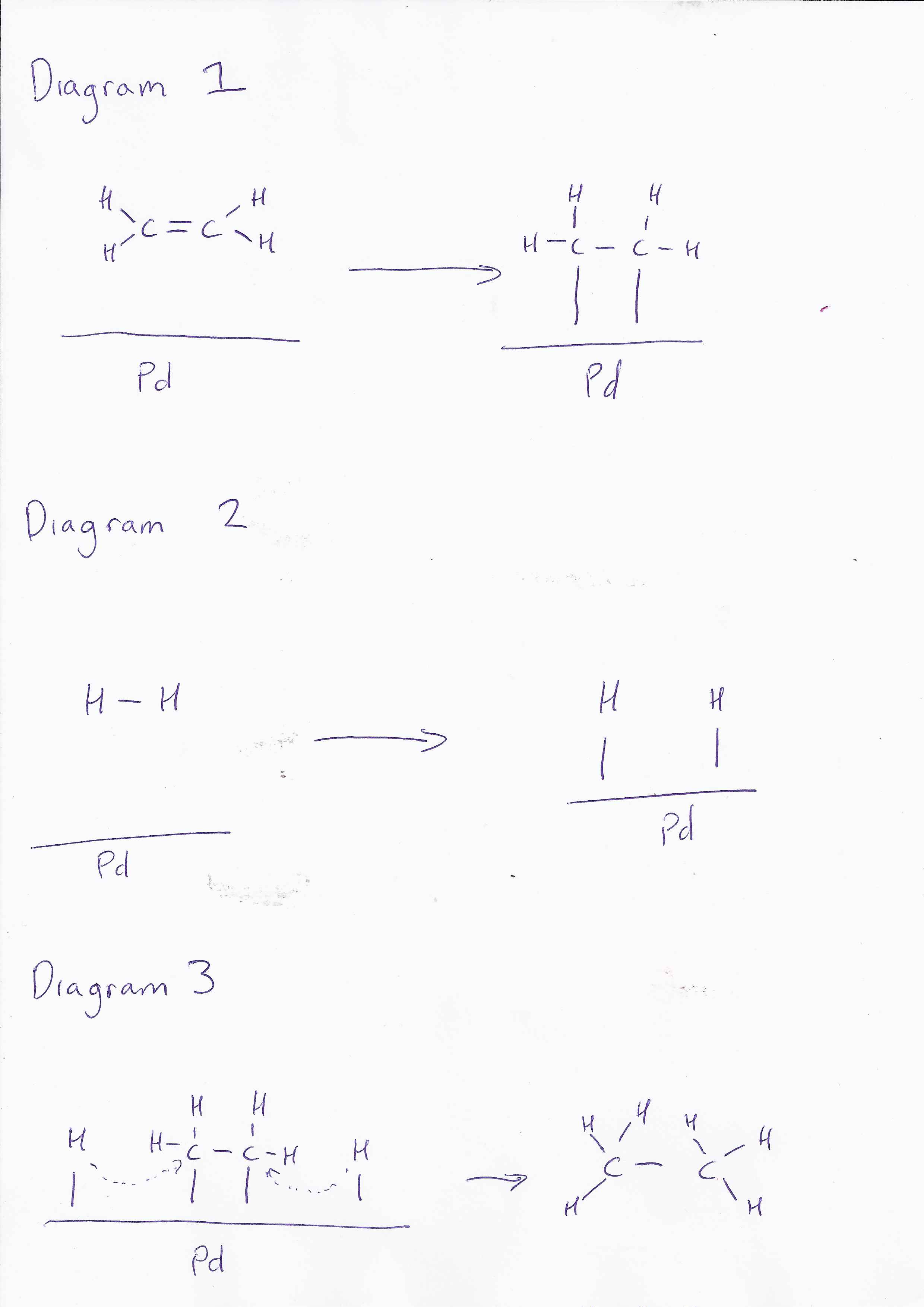
Um... I'll take a stab at simplifying for you- but it's not really my area (only think i've seen in detail recently was Pd chemistry... and I forget most of that lol). Ok, so- imagine you have a Pd surface- it is a metal, so there are electrons delocalised over the entire surface. Next you have an ethylene molecule- its C=C bond is electron rich, and has easy to access pi- electrons. The pi orbitals interact with the Pd surface and the electrons in the two systems can interact. Diagramatically (I hope the spaces come out): Diagram 1 So the new bonds between the carbon and palladium are weaker than the pi bond. At the same time, Pd has an affinity for H in much the same way: Diagram 2 The Pd surface now has C2H4 and 2H clusters all positioned relatively close to each other- and the final reaction can proceed forming ethane: Diagram 3 So, the energy of the total reaction is the same, but the initial energy required is less because the energy to form the transition state is less. As to why this is less, I will let someone else pick up on it- as I'm not all that sure myself (though I have a few ideas). Edit: Spaces didn't work so included pretty picture!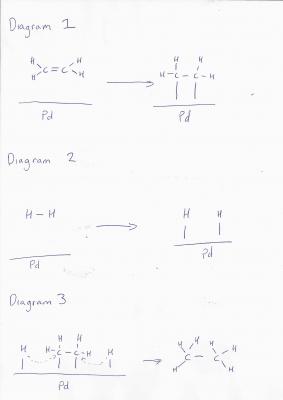
-
No idea. Petrol contains a lot of things though- no doubt a small percentage is tBuOH. But there's got to be an easier way... You could probably react methyl lithium or methyl magnesium bromide with acetone, then acid work up. Its just a bit tedious and expensive for something that you would probably want in solvent quantities (i.e. 100s of grams).
-
Easiest way is CaCl2 and MgSO4. These are both readily available- any CaSO4 still present can be removed by boiling and filtering. Edit: Just in case it wasn't clear, the CaSO4 is a significantly insoluble salt and precipitates removing it from solution. You need a 1:1 molar ratio.
-
Oh, also I missed a bit about reactivity in my above post (I was a little hurried). Primary alcohols undergo a reaction known as SN2 where a nucleophile (say cyanide CN-) attacks the alcoholic carbon from the opposite side of the OH group. As the CN- gets closer the OH gets further away and eventually leaves as OH-. Secondary alcohols can also undergo this mechanism (but at a reduced rate due to steric hinderance). Tertiary alcohols are too hindered for this mechanism to work, and instead the OH- leaves first (because the carbo-cation is stable enough to remain independantly). When the CN- encounters the carbocation a the new compound forms (however the product is racemic). Again secondary alcohols can also react in this way. As such, tertiary alcohols are more reactive as the rate limiting step is the first step dependant on carbocation stability. Secondary alcohols usually follow (depending on the stability of the carbocation formed) and primary alcohols, having no stable carbocations, are soley rate dependant on the concentration of CN- (as well as to drive the reaction forward) which is available to 'attack'.